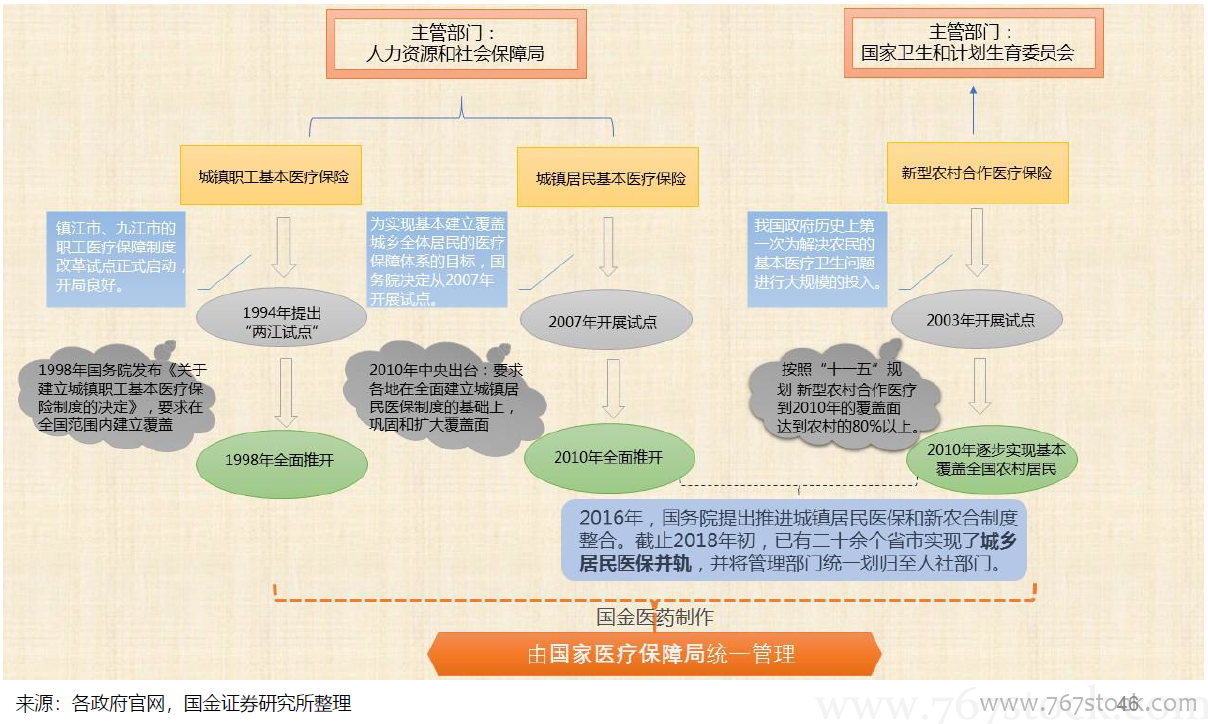
国务院关于改革药品医疗器械审评审批制度的意见(“44号文”)。《意见》明确了改革的12项任务,包括提高药品审批质量,解决注册积压,推进仿制药质量一致性评价,加快创新药的审评审批,开展药品上市许可持有人制度试点,落实申请人申报主体责任,及时发布药品供求和申报信息(限制性清单、鼓励性清单),改进药品临床试验审批,严肃查处注册申报弄虚作假行为,简化药品审批程序、完善药品再注册制度,改革医疗器械审批方式,健全审评质量控制体系,全面公开药品医疗器械审评审批信息。
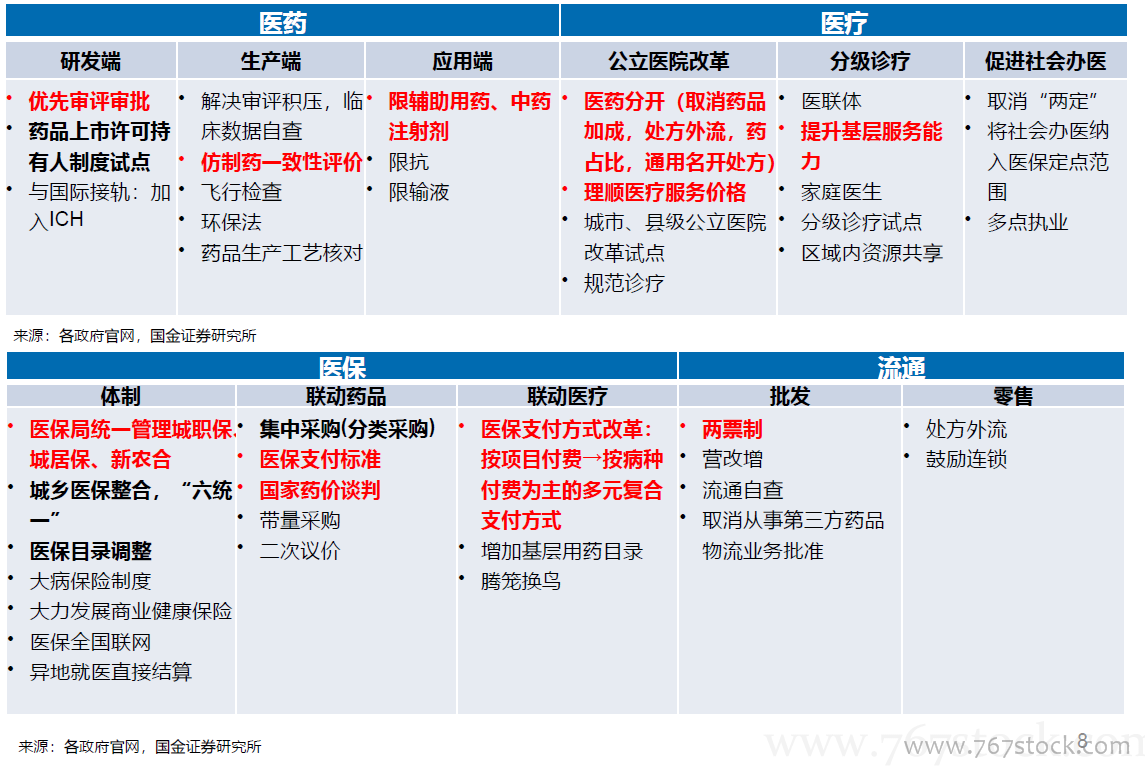 总局关于药品注册审评审批若干政策的公告(“230号公告”)。
总局关于药品注册审评审批若干政策的公告(“230号公告”)。10大政策:提高仿制药审批标准;规范改良型新药的审评审批;优化临床试验申请的审评审批:BE由审批制改为备案制;实行同品种集中审评:对已经受理的相同品种,按照统一的审评标准和尺度组织力量进行集中审评;允许申请人主动撤回不符合条件的药品注册申请;严格审查药品的安全性和有效性;加快临床急需等药品的审批:符合条件的可向CDE申请;严惩临床试验数据造假行为;引导申请人理性申报;规范药品注册复审工作。
 总局办公厅公开征求《药品注册管理办法(修订稿)》意见。
总局办公厅公开征求《药品注册管理办法(修订稿)》意见。全面落实上市许可持有人制度;审评审批时限更明确:将药品注册的受理、审查、审批各环节的时间限制专门列出一个章节。
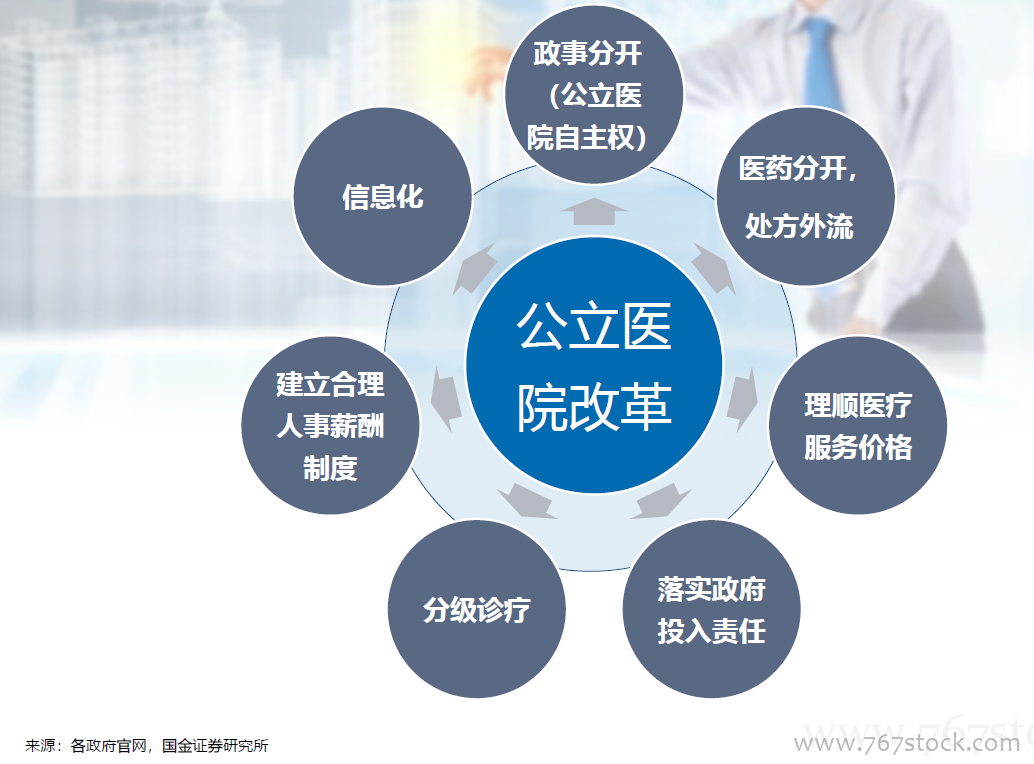 〈中华人民共和国药品管理法〉修正案(草案征求意见稿)。
〈中华人民共和国药品管理法〉修正案(草案征求意见稿)。全面补充MAH内容:国家实行药品上市许可持有人制度,药品上市许可持有人对药品安全、有效和质量可控承担法律责任。审批时间:国务院药品监督管理部门应当自受理新药临床试验申请之日起60个工作日内决定是否同意开展临床试验;逾期未通知的,申请人可以开展临床试验。落实处罚到人要求:对数据造假、药品安全等明确惩罚措施。纳入CRO为药品研发责任人,对违规方采取重罚措施;落实行政审批制度改革要求:临床试验机构由认证改为备案,药物临床试验审批由明示许可改为默示许可,生物等效性试验实行备案管理,取消药品生产质量管理规范认证、药品经营质量管理规范认证制度,将原料药和辅料修改为与药品一并审批。
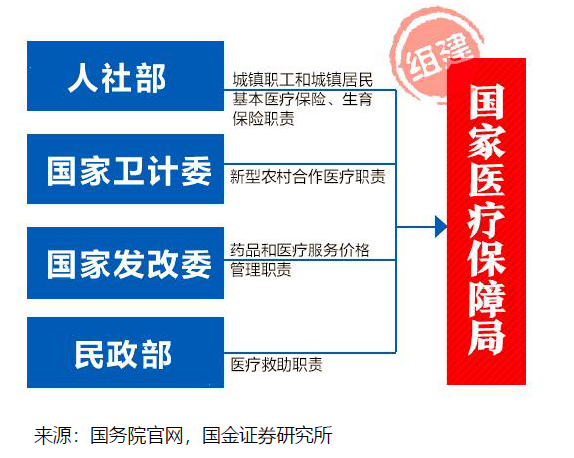 关于深化审评审批制度改革鼓励药品医疗器械创新的意见。
关于深化审评审批制度改革鼓励药品医疗器械创新的意见。改革临床试验管理:增加临床试验产能,加快临床试验和审评流程,关注CRO、海外数据转报和创新药企业 加快上市审评审批:对临床急需、罕见病有条件批准上市、优先审评。促进药品创新和仿制药发展:完善上市药品信息监管,鼓励仿制药专利挑战,加大创新药的生命周期保护和医保支持。
【免费下载】
http://tinyurl.com/y7qjz84p
【备用下载】


 扫码加好友,拉您进群
扫码加好友,拉您进群 全部版块
全部版块 我的主页
我的主页

 收藏
收藏






